

Faits sur les superoxydes dismutases : Rôle dans la signalisation des oxydes rouges
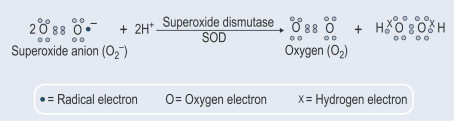
Les SOD représentent le premier système de défense enzymatique contre les dommages radicaux causés par l'oxygène : ainsi, cette enzyme est essentielle pour tous les organismes aérobies, mais pas pour les anaérobies. À l'appui de cette hypothèse, McCord pense que l'existence d'un organisme aérobie dépend principalement de sa capacité à produire des SOD puisque sa carence est responsable de la sensibilité à l'oxygène et ne permet de survivre qu'en milieu anaérobie.
Dans des conditions physiologiques, les superoxydes dismutases, ainsi que les piégeurs non enzymatiques ROS comme les vitamines E, A et C, maintiennent un état stable entre les systèmes oxydant et antioxydant (Russo et al., 2011). La dysrégulation de l'homéostasie redox, déterminée par un déséquilibre entre la production de ROS et la capacité de piégeage, entraîne des dommages cellulaires considérables tels que la lipoperoxydation membranaire, l'acide nucléique et les altérations structurelles des protéines contribuant aux maladies neurodégénératives et cardiovasculaires.
Ces dernières années, de nombreuses données obtenues lors d'études in vitro réalisées sur de nombreuses lignées cellulaires, principalement des cellules de neuroblastome SK-N-BE, indiquent que la SOD1 est sécrétée et est capable d'activer, par le biais du récepteur muscarinique M1, des voies cellulaires impliquant l'activation des ERK1/2 et des AKT ; ces effets sont associés à une augmentation du calcium intracellulaire qui est encore accentuée lorsque ces cellules sont stimulées par la SOD1G93A mutée.
La localisation intracellulaire cytosolique de la SOD1 a fait l'objet d'un débat ; des preuves récentes, réalisées sur des cellules de neuroblastome neuro2 de souris transfectées, ont démontré que la SOD1 de type sauvage (wt-SOD1) et les mutants de la SOD1 sont distribués dans les structures lumineuses de l'appareil endoplasmique et de Golgi (Urushitani et al., 2008). Les premières preuves expérimentales que certaines lignées cellulaires pourraient être capables de sécréter la superoxyde dismutase Cu,Zn remontent à de nombreuses années, lorsque nous avons, pour la première fois, montré la sécrétion de cette protéine par des expériences réalisées dans des hépatocytes et des fibroblastes (Mondola et al., 1996), des cellules de neuroblastome SK-N-BE (Mondola et al., 1998 ; Gomes et al., 2007 ; Polazzi et al., 2013) et dans des cellules épithéliales dérivées du thymus (Cimini et al., 2002).
En outre, nous avons démontré que dans les cellules de neuroblastome humain SK-N-BE, qui présentent une plus grande sensibilité à la cytotoxicité induite par la privation de glucose en raison d'une sensibilité accrue aux ROS (Shutt et al., 2010), l'exportation de SOD1 a lieu dans des conditions normales et est accrue à la suite d'un stress oxydatif (Mondola et al., 1996, 1998). Nous avons montré successivement que la SOD1 des cellules de neuroblastome humain SK-N-BE est exportée par une voie de sécrétion microvésiculaire qui est altérée par la breeldine A (BFA), et par le 2-désoxyglucose, et l'azide de sodium, qui réduit le pool intracellulaire d'ATP (Mondola et al., 2003).
Indicateurs sur le rôle de la superoxyde dismutase de manganèse dans ... - Hindawi You Need To Know

Un autre aspect important a été la découverte qu'outre l'exportation de la SOD1 constitutive, la sécrétion de cette enzyme est également induite. À cet égard, nous avons montré (Santillo et al., 2007) que la SOD1 est activement libérée des synaptosomes du cerveau du rat ainsi que des cellules GH3 de l'hypophyse du rat qui représentent un bon modèle pour étudier la libération inductible de la SOD1 puisqu'elles possèdent toute la machinerie protéique neuronale impliquée dans l'exocytose des vésicules synaptiques.
En outre, dans le but d'évaluer le rôle possible joué par l'exportation de SOD1, nous avons récemment démontré, dans la lignée cellulaire de neuroblastome SK-N-BE, que cette enzyme est capable, grâce à l'implication du récepteur muscarinique M1, d'activer l'ERK1/2 et l'AKT en fonction de la dose et du temps. Cet effet a été remarquablement réduit par la mise au silence du récepteur M1 ainsi que par l'utilisation de la pirenzépine, un antagoniste M1 (Damiano et al., 2013).
Cependant, il a été démontré que le FGF-1 et l'isoforme de 18 kDa du FGF-2 sont sécrétés par une voie alternative directement transloquée du cytoplasme dans l'espace extracellulaire. De même, l'interleukine 1β (IL-1β) a été signalée comme étant sécrétée par une voie d'exportation vésiculaire non classique. Les protéines solubles contiennent classiquement des peptides signaux N-terminaux qui les dirigent vers l'appareil de translocation du Réticulum Endoplasmique (RE) (Walter et al., 1984).
En outre, la sécrétion de protéines non classiques dépend à la fois de l'énergie et de la température et peut être stimulée ou inhibée par divers traitements (Cleves, 1997 ; Hughes, 1999). La liste des protéines qui pourraient être exportées des cellules en l'absence d'un système ERG fonctionnel (voie de sécrétion non conventionnelle), comme l'IL-1β et la galectine 1 (également appelée L-14), ne cesse de s'allonger ; pour d'autres données, voir la revue de Nickel (2003).

Pour cette raison, la sécrétion de la SOD1 devrait contourner la voie de sécrétion canonique de l'ERG. Nous avons précédemment démontré que le BFA ainsi que le 2-désoxyglucose et l'azide de sodium (NaN3), entravent l'exportation de la SOD1 (Mondola et al., 2003). À notre avis, le traitement à l'acide folique ne régule probablement pas seulement la voie sécrétoire classique de l'ERG, mais aussi le trafic de la membrane microvésiculaire de la sécrétion protéique non conventionnelle ou de l'exportation alternative de protéines.
L'astuce de 20 secondes pour la superoxyde dismutase : avantages pour la santé, utilisations, effets secondaires ...
De plus, ces auteurs ont montré que le SOD1A4V inhibe le transport des protéines sécrétoires des Urgences vers l'appareil de Golgi. La sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative d'apparition adulte, caractérisée par la mort sélective des motoneurones supérieurs et inférieurs du cerveau et de la moelle épinière. Les symptômes comprennent l'atrophie musculaire, la spasticité, la paralysie et la mort par insuffisance respiratoire dans les 3 à 5 ans suivant le diagnostic.
Les processus cliniques et pathologiques indiquent que le stress des urgences représente une voie clé impliquée dans la mort cellulaire. Dans le modèle transgénique SOD1G93A de SLA chez le rat, une réponse protéique dépliée et une apoptose induite par le stress ER ont été observées (Atkin et al., 2006) ; une réponse protéique dépliée, comprenant l'induction de kinases de capteurs de stress, de chaperons et de médiateurs apoptotiques, a également été démontrée dans les motoneurones de la moelle épinière de patients humains atteints de la forme sporadique de SLA (sALS) qui n'est pas limitée aux mutations SOD1 (Atkin et al., 2008).
La SOD1 et d'autres protéines sont mal repliées dans l'EVAA et dans l'ALS, mais on ne sait pas exactement comment cela déclenche le stress du RE, la fragmentation de l'appareil de Golgi, la perturbation du transport axonal et l'apoptose. Près de 20% de l'EVAA est causée par des mutations du gène SOD1 (Neumann et al., 2006). En effet, la majorité des mutants SOD1 conservent leur activité enzymatique, ce qui suggère l'apparition d'une fonction de gain d'activité toxique plutôt qu'une simple perte de fonction (Strong et al., 2005 ; Dion et al., 2009).
(2005) ont démontré une altération de la sécrétion extracellulaire constitutive de la SOD1 mutante dans les cellules NSC-34 qui induit de fréquentes inclusions cytoplasmiques et une insolubilité des protéines. Ces données établissent un lien entre la sécrétion déficiente de la SOD1 mutante et les agrégats protéiques intracellulaires et la toxicité dans les cellules NSC-34. En outre, ces auteurs ont montré que dans un modèle de SLA chez le rat transgénique, la perfusion intraspinale chronique de wt-SOD1 exogène humain retardait de manière significative la progression de la maladie, ce qui suggère un nouveau rôle extracellulaire pour la SOD1 dans la SLA ; par conséquent, la délivrance extracellulaire de wt-SOD1 humain pourrait améliorer la maladie clinique chez les rats transgéniques atteints de SLA, ce qui confirme le nouveau rôle extracellulaire de la SOD1 mutante et de la wt-SOD1 dans la pathogenèse et la thérapie de la SLA, respectivement.
En outre, chez des souris transgéniques, porteuses de mutations SOD1, des effets toxiques sur les motoneurones par activation de la microglie ont été observés (Urushitani et al., 2006 ; Zhao et al., 2010). Les cellules microgliales peuvent jouer un rôle important dans la progression de la SLA (Pramatarova et al., 2001) puisque l'activation des microglies peut être observée avant la perte de neurones chez les souris transgéniques exprimant des mutants SOD1 humains (Alexianu et al., 2001).
Merci de partager vos informations. J'apprécie vraiment vos efforts et j'attends vos prochains remerciements. Tandie Lazarus Hughes